---
title: "Publish Ichthyoplankton to OBIS"
calcofi:
target_name: publish_ichthyo_to_obis
workflow_type: publish
dependency:
- release_database
output: data/darwincore/ichthyo_*.zip
editor_options:
chunk_output_type: console
---
## Overview
This workflow converts CalCOFI ichthyoplankton (fish eggs and larvae) data to an OBIS-compliant DarwinCore Archive for publication. The archive follows the Event Core structure with Occurrence and ExtendedMeasurementOrFact (eMoF) extensions.
**Note on bottle data**: CalCOFI bottle hydrography (temperature, salinity, dissolved oxygen, nutrients, chlorophyll, carbon system parameters) is not included in this DarwinCore Archive. These environmental data are already served via [ERDDAP CalCOFI SIO Hydrographic Bottle Data](https://coastwatch.pfeg.noaa.gov/erddap/tabledap/siocalcofiHydroBottle.html).
**Data source**: Frozen CalCOFI database release on GCS via `calcofi4r::cc_get_db()`, containing:
- SWFSC ichthyoplankton tables (cruise, ship, site, tow, net, ichthyo, species, lookup, taxon, taxa_rank)
**References**: [CalCOFI Fish Eggs & Larvae](https://calcofi.org/data/marine-ecosystem-data/fish-eggs-larvae/) | [OBIS Manual](https://manual.obis.org/) | [DarwinCore Terms](https://dwc.tdwg.org/terms/)
```{r}
#| label: setup
devtools::load_all(here::here("../calcofi4db"))
librarian::shelf(
CalCOFI / calcofi4db,
DBI,
dplyr,
DT,
duckdb,
EML,
glue,
here,
lubridate,
iobis / obistools,
purrr,
stringr,
readr,
tibble,
tidyr,
uuid,
xml2,
quiet = T
)
options(readr.show_col_types = F)
# calcofi namespace for deterministic UUID v5 generation
CALCOFI_NS <- "c0f1ca00-ca1c-5000-b000-1c4790000000"
# dataset metadata ----
dataset_short_name <- "calcofi_ichthyo"
dataset_title <- "CalCOFI Fish Larvae & Egg Tows"
dataset_abstract <- "Fish larvae counts and standardized counts for eggs captured in CalCOFI ichthyoplankton nets (primarily vertical [Calvet or Pairovet], oblique [bongo or ring nets], and surface tows [Manta nets]). Surface tows are normally standardized to count per 1,000 m^3 strained. Oblique tows are normally standardized to count per 10 m^2 of surface sampled."
dataset_url <- "https://calcofi.org/data/marine-ecosystem-data/fish-eggs-larvae/"
dataset_logo_url <- "https://calcofi.org/wp-content/uploads/2021/03/cropped-calcofirose_512_favicon.png"
dataset_keywords <- c(
"atmosphere",
"biology",
"biosphere",
"calcofi",
"earth science",
"environment",
"ichthyoplankton",
"latitude",
"longitude",
"ocean",
"time"
)
country_code <- "US"
water_body <- "California Current"
dataset_id <- glue(
"calcofi.io_workflows_ichthyo_to_obis_{format(Sys.Date(), '%Y-%m-%d')}"
)
sampling_methods_description <- "Standard CalCOFI ichthyoplankton tows. The standard oblique tow uses a bongo net (71 cm diameter, 0.505 mm mesh) or 1-m ring net (prior to 1978) retrieved at a constant wire angle (45 degrees) from 210 m depth to surface. Surface tows use a Manta net; vertical tows use CalVET/Pairovet nets. Flowmeters measure volume filtered. Samples are preserved in 5% formalin. In the lab, fish eggs and larvae are sorted, identified to lowest taxon possible, enumerated, and measured."
study_extent_description <- "The study covers the California Current ecosystem, primarily off Southern California (standard lines 77-93) but historically extending from the border of Canada to the tip of Baja California. The time series for this dataset generally begins in 1951 and continues to the present, with quarterly cruises."
sampling_description <- "Samples are collected at fixed stations along the CalCOFI grid. Oblique tows are standardized to counts per 10 m^2 of sea surface. Surface tows are standardized to counts per 1,000 m^3. Data includes raw counts (tallies) and standardized abundances."
funding_information <- "CalCOFI is a partnership between the NOAA National Marine Fisheries Service (NMFS), Scripps Institution of Oceanography (SIO), and California Department of Fish and Wildlife (CDFW)."
rights <- "The data may be used and redistributed for free but is not intended for legal use, since it may contain inaccuracies. Neither the data Contributor, ERD, NOAA, nor the United States Government, nor any of their employees or contractors, makes any warranty, express or implied, including warranties of merchantability and fitness for a particular purpose, or assumes any legal liability for the accuracy, completeness, or usefulness, of this information."
license <- "To the extent possible under law, the publisher has waived all rights to these data and has dedicated them to the Public Domain (CC0 1.0). Users may copy, modify, distribute and use the work, including for commercial purposes, without restriction."
license_xml <- c(
"Public Domain (CC0 1.0)" = '<ulink url="http://creativecommons.org/publicdomain/zero/1.0/legalcode"><citetitle>Public Domain (CC0 1.0)</citetitle></ulink>'
)
intellectual_rights <- list(
para = paste(rights, license, collapse = " ")
)
dir_out <- here("data/darwincore/ichthyo")
# connect to latest frozen release on GCS ----
con <- calcofi4r::cc_get_db()
```
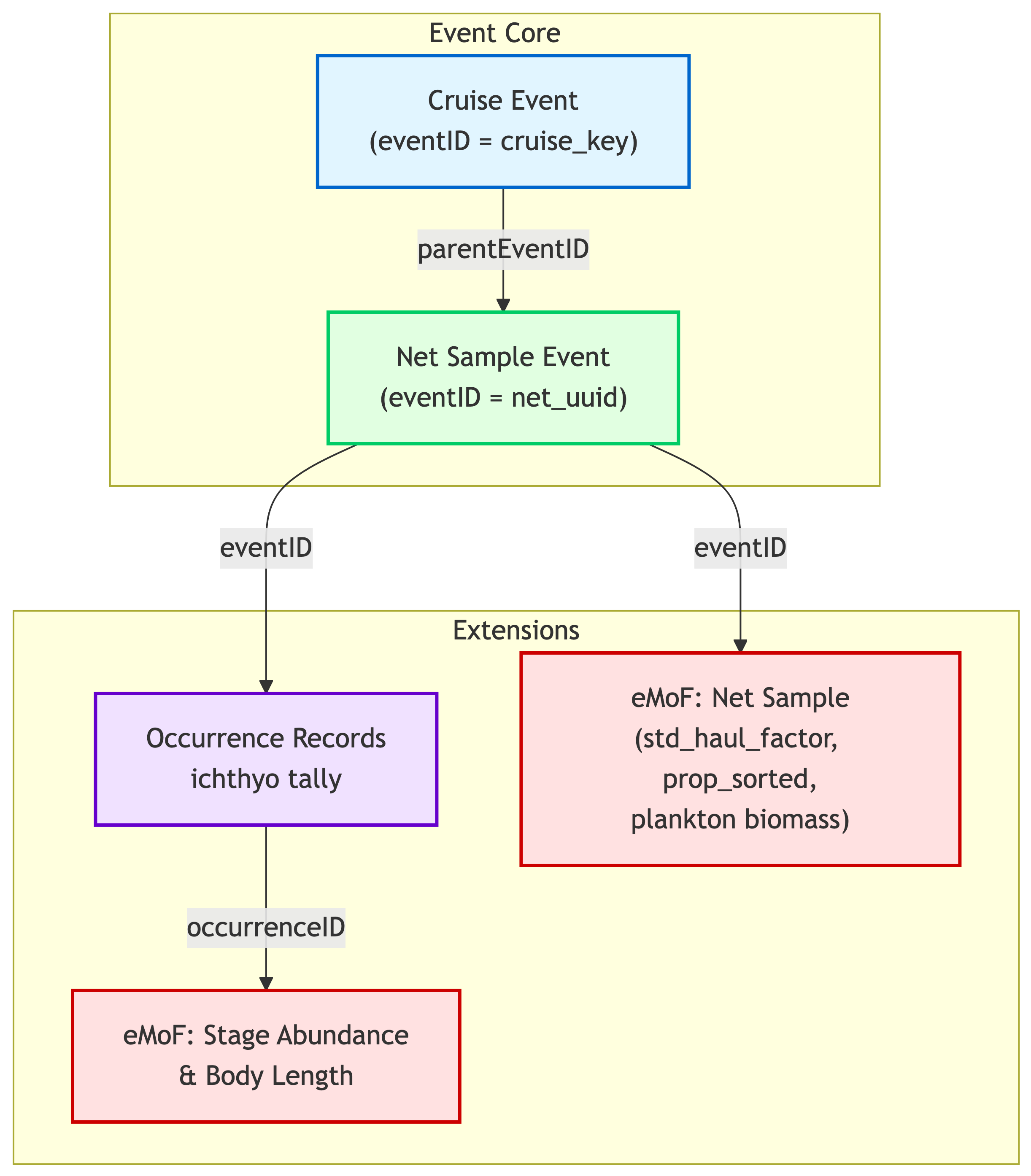
## Event Hierarchy Design
The archive uses a **two-level event hierarchy**: ichthyoplankton net tows are children of cruise events. Each net sample carries its own coordinates and timestamp.
```{mermaid}
%%| label: fig-ichthyo_event_hierarchy
%%| fig-cap: "Event hierarchy for CalCOFI ichthyoplankton data in DarwinCore Archive format."
%%| file: diagrams/ichthyo_event_hierarchy.mmd
```
**Key design decisions**:
1. **Collapsed hierarchy**: cruise → net_sample (no intermediate site/tow levels) to avoid event inheritance issues with NA values in the IPT
2. **`locationID`**: net_sample events use `locationID = "{line}_{station}"` for spatial joining by OBIS consumers
3. **Occurrence consolidation**: one occurrence per life stage (egg or larva) per species per net sample
4. **eMoF structure**: three sub-tables covering net-level measurements, stage abundance, and body length
## Data Extraction
Load all necessary tables from the merged DuckDB:
```{r}
#| label: extract-tables
# ichthyo chain ----
tbl_cruise <- tbl(con, "cruise")
tbl_ship <- tbl(con, "ship")
# use SQL to exclude GEOMETRY column (unsupported by duckdb R driver)
site_cols <- dbGetQuery(con,
"SELECT column_name FROM information_schema.columns
WHERE table_name = 'site' AND data_type NOT LIKE 'GEOMETRY%'")$column_name
tbl_site <- tbl(con, sql(
glue("SELECT {paste(site_cols, collapse = ', ')} FROM site")))
tbl_tow <- tbl(con, "tow")
tbl_net <- tbl(con, "net")
tbl_ichthyo <- tbl(con, "ichthyo")
tbl_species <- tbl(con, "species")
tbl_lookup <- tbl(con, "lookup")
# taxonomy ----
tbl_taxon <- tbl(con, "taxon")
tbl_taxa_rank <- tbl(con, "taxa_rank")
# quick counts (use SQL to avoid GEOMETRY column issues)
cat("Table row counts:\n")
for (tname in c(
"cruise", "ship", "site", "tow", "net",
"ichthyo", "species", "lookup", "taxon", "taxa_rank"
)) {
n <- dbGetQuery(con, glue("SELECT COUNT(*) AS n FROM {tname}"))$n
cat(glue(" {tname}: {prettyNum(n, big.mark = ',')}"), "\n")
}
```
## Collect Tables with Unique Identifiers
The merged DuckDB uses `*_uuid` columns as primary keys for SWFSC tables
(site, tow, net, ichthyo). These UUIDs are already globally unique and
stable — `net_uuid` is minted at sea and `ichthyo_uuid` is a deterministic
UUID v5 from the composite natural key — so no additional minting is needed.
```{r}
#| label: collect-uuids
# collect net table with UUIDs for eventID ----
# as.UUID() coerces to native 128-bit UUID class (always lowercase,
# faster comparisons than character strings)
d_net <- tbl_net |>
collect() |>
mutate(across(ends_with("_uuid"), as.UUID))
stopifnot(all(!duplicated(d_net$net_uuid)))
cat(glue("Net UUIDs: {nrow(d_net)} (all unique)"), "\n")
# collect ichthyo table with UUIDs ----
d_ichthyo <- tbl_ichthyo |>
collect() |>
mutate(across(ends_with("_uuid"), as.UUID))
stopifnot(all(!duplicated(d_ichthyo$ichthyo_uuid)))
cat(glue("Ichthyo UUIDs: {nrow(d_ichthyo)} (all unique)"), "\n")
```
## Life Stage Vocabulary
Define mappings for egg and larva developmental stages to descriptive terms:
```{r}
#| label: vocabulary-lookups
# P06 measurement unit vocabulary (NERC) ----
measurement_unit_vocab <- tibble(
measurementUnit = c(
"individuals",
"millimeters",
"dimensionless",
"cubic meters",
"grams",
"proportion",
"m^3",
"meters"
),
measurementUnitID = c(
"http://vocab.nerc.ac.uk/collection/P06/current/UUUU/", # individuals
"http://vocab.nerc.ac.uk/collection/P06/current/UXMM/", # millimeters
"http://vocab.nerc.ac.uk/collection/P06/current/UUUU/", # dimensionless
"http://vocab.nerc.ac.uk/collection/P06/current/MCUB/", # cubic meters
"http://vocab.nerc.ac.uk/collection/P06/current/UGRM/", # grams
"http://vocab.nerc.ac.uk/collection/P06/current/UUUU/", # proportion
"http://vocab.nerc.ac.uk/collection/P06/current/MCUB/", # m^3
"http://vocab.nerc.ac.uk/collection/P06/current/ULAA/" # meters
)
)
```
## Build Event Hierarchy
### Cruise Events
Aggregate over net tows to build cruise-level events:
```{r}
#| label: build-cruise-events
# collect joined net→tow→site for cruise summaries ----
d_net_site <- d_net |>
left_join(
tbl_tow |>
collect() |>
mutate(across(ends_with("_uuid"), as.UUID)) |>
select(tow_uuid, site_uuid, time_start),
by = "tow_uuid"
) |>
left_join(
tbl_site |>
collect() |>
mutate(across(ends_with("_uuid"), as.UUID)) |>
select(site_uuid, cruise_key, longitude, latitude, line, station),
by = "site_uuid"
)
# net cruise summaries ----
d_net_cruise_summary <- d_net_site |>
group_by(cruise_key) |>
summarize(
net_time_min = min(time_start, na.rm = TRUE),
net_time_max = max(time_start, na.rm = TRUE),
net_lon_min = min(longitude, na.rm = TRUE),
net_lon_max = max(longitude, na.rm = TRUE),
net_lat_min = min(latitude, na.rm = TRUE),
net_lat_max = max(latitude, na.rm = TRUE),
.groups = "drop"
)
# combine cruise metadata ----
d_cruise_events <- tbl_cruise |>
left_join(tbl_ship, by = "ship_key") |>
collect() |>
left_join(d_net_cruise_summary, by = "cruise_key") |>
mutate(
# date range from net tows
time_min = net_time_min,
time_max = net_time_max,
# bounding box from net sites
lon_min = net_lon_min,
lon_max = net_lon_max,
lat_min = net_lat_min,
lat_max = net_lat_max
) |>
filter(!is.na(time_min)) |>
mutate(
eventID = cruise_key,
parentEventID = NA_character_,
eventType = "cruise",
eventDate = case_when(
!is.na(time_min) & !is.na(time_max) ~
paste0(
format(time_min, "%Y-%m-%dT%H:%M:%SZ"),
"/",
format(time_max, "%Y-%m-%dT%H:%M:%SZ")
),
TRUE ~ NA_character_
),
footprintWKT = case_when(
!is.na(lon_min) ~
glue(
"POLYGON(({lon_min} {lat_min}, {lon_max} {lat_min}, {lon_max} {lat_max}, {lon_min} {lat_max}, {lon_min} {lat_min}))"
),
TRUE ~ NA_character_
),
samplingProtocol = "Marine plankton survey cruise",
habitat = "marine",
eventRemarks = glue(
"Cruise on {ship_name} from {as.Date(time_min)} to {as.Date(time_max)}"
)
) |>
select(
eventID,
parentEventID,
eventType,
eventDate,
footprintWKT,
samplingProtocol,
habitat,
eventRemarks
)
cat(glue("Cruise events: {nrow(d_cruise_events)}"), "\n")
```
### Net Sample Events
Collapsed net_sample events (cruise → net_sample) with coordinates, date, and sampling info:
```{r}
#| label: build-net-events
d_net_events <- d_net_site |>
mutate(
eventID = as.character(net_uuid), # <-- cast to allow cruise_key as another eventID type
parentEventID = cruise_key,
eventType = "net_sample",
eventDate = format(time_start, "%Y-%m-%dT%H:%M:%SZ"),
decimalLatitude = latitude,
decimalLongitude = longitude,
locationID = paste0(line, "_", station),
samplingProtocol = "CalCOFI ichthyoplankton net tow",
sampleSizeValue = vol_sampled_m3,
sampleSizeUnit = "m^3",
eventRemarks = glue(
"Net: {side} side; ",
"{prop_sorted} proportion sorted of {vol_sampled_m3} m3 sampled"
)
)
# keep full data for eMoF
d_net_events_full <- d_net_events
d_net_events <- d_net_events |>
select(
eventID,
parentEventID,
eventType,
eventDate,
decimalLatitude,
decimalLongitude,
locationID,
samplingProtocol,
sampleSizeValue,
sampleSizeUnit,
eventRemarks
)
stopifnot(all(!duplicated(d_net_events$eventID)))
cat(glue("Net sample events: {nrow(d_net_events)}"), "\n")
```
### Combine All Events
```{r}
#| label: combine-events
d_event <- bind_rows(
d_cruise_events,
d_net_events
) |>
mutate(
geodeticDatum = "WGS84",
coordinateUncertaintyInMeters = 1000,
countryCode = country_code,
waterBody = water_body,
datasetID = dataset_id
)
cat("Event counts by type:\n")
d_event |> count(eventType) |> print()
# verify all child events have valid parentEventID
cruise_ids <- d_event |> filter(eventType == "cruise") |> pull(eventID)
child_events <- d_event |> filter(eventType != "cruise")
orphan_children <- child_events |> filter(!parentEventID %in% cruise_ids)
if (nrow(orphan_children) > 0) {
warning(glue(
"{nrow(orphan_children)} child events have no matching cruise parent"
))
} else {
cat("All child events have valid cruise parents.\n")
}
```
## Build Occurrence Records
From the consolidated `ichthyo` table — one occurrence per life stage per species per net sample (tally rows where `measurement_type` is NULL):
```{r}
#| label: build-occurrences
d_occurrence <- d_ichthyo |>
filter(is.na(measurement_type)) |>
left_join(
tbl_species |> collect(),
by = "species_id"
) |>
mutate(
occurrenceID = as.character(ichthyo_uuid),
eventID = as.character(net_uuid),
scientificName = scientific_name,
scientificNameID = ifelse(
!is.na(worms_id) & worms_id > 0,
paste0("urn:lsid:marinespecies.org:taxname:", worms_id),
NA_character_
),
kingdom = "Animalia",
occurrenceStatus = ifelse(tally > 0, "present", "absent"),
organismQuantity = tally,
organismQuantityType = "individuals",
lifeStage = life_stage,
preparations = paste0(life_stage, " sample"),
basisOfRecord = "HumanObservation",
modified = Sys.Date()
)
# verify all occurrences link to valid net_sample events
net_ids <- d_net_events |> pull(eventID)
orphan_occs <- d_occurrence |> filter(!eventID %in% net_ids)
if (nrow(orphan_occs) > 0) {
warning(glue(
"{nrow(orphan_occs)} occurrences have no matching net_sample event"
))
} else {
cat("All occurrences link to valid net_sample events.\n")
}
d_occurrence_out <- d_occurrence |>
select(
occurrenceID,
eventID,
scientificName,
scientificNameID,
kingdom,
occurrenceStatus,
organismQuantity,
organismQuantityType,
lifeStage,
preparations,
basisOfRecord,
modified
)
cat(glue("Occurrences: {nrow(d_occurrence_out)}"), "\n")
cat(glue("Unique species: {n_distinct(d_occurrence_out$scientificName)}"), "\n")
```
## Build ExtendedMeasurementOrFact Extension
### 10a. Net-level sample measurements
```{r}
#| label: emof-net-level
d_emof_net <- d_net_events_full |>
select(
eventID,
net_uuid,
std_haul_factor,
prop_sorted,
smallplankton,
totalplankton
) |>
pivot_longer(
cols = c(std_haul_factor, prop_sorted, smallplankton, totalplankton),
names_to = "meas_type",
values_to = "measurementValue"
) |>
filter(!is.na(measurementValue)) |>
mutate(
measurementID = UUIDfromName(
CALCOFI_NS,
paste0("net:", net_uuid, ":", meas_type)
),
occurrenceID = NA_character_,
measurementType = case_when(
meas_type == "std_haul_factor" ~ "standardized haul factor",
meas_type == "prop_sorted" ~ "proportion of sample sorted",
meas_type == "smallplankton" ~ "small plankton biomass",
meas_type == "totalplankton" ~ "total plankton biomass"
),
measurementTypeID = NA_character_,
measurementUnit = case_when(
meas_type == "std_haul_factor" ~ "dimensionless",
meas_type == "prop_sorted" ~ "dimensionless",
meas_type %in% c("smallplankton", "totalplankton") ~ "grams"
),
measurementMethod = "https://oceanview.pfeg.noaa.gov/CalCOFI/calcofi_info.html",
measurementRemarks = case_when(
meas_type == "std_haul_factor" ~
"Standardization factor for water filtered; abundances per 10 m^2 (Smith 1977)",
meas_type == "prop_sorted" ~ "Fraction of total sample examined",
TRUE ~ NA_character_
)
) |>
left_join(measurement_unit_vocab, by = "measurementUnit") |>
select(
eventID,
occurrenceID,
measurementID,
measurementType,
measurementTypeID,
measurementValue,
measurementUnit,
measurementUnitID,
measurementMethod,
measurementRemarks
)
cat(glue("eMoF net-level: {nrow(d_emof_net)}"), "\n")
```
### 10b. Stage-specific abundance
From `ichthyo` rows where `measurement_type = 'stage'`, linked to the matching tally row's `ichthyo_uuid` as `occurrenceID`:
```{r}
#| label: emof-stage-abundance
# build lookup: tally occurrenceIDs with their eventID (net_uuid)
d_occ_lookup <- d_occurrence |>
select(
net_uuid,
species_id,
life_stage,
occurrenceID = ichthyo_uuid,
occ_eventID = eventID
) |>
mutate(occurrenceID = as.character(occurrenceID))
# collect lookup descriptions for stages
d_lookup <- tbl_lookup |>
filter(lookup_type %in% c("egg_stage", "larva_stage")) |>
collect()
# stage records — join to lookup via derived lookup_type
d_emof_stage <- d_ichthyo |>
filter(measurement_type == "stage") |>
mutate(
lookup_type = paste0(life_stage, "_stage")
) |>
left_join(
d_lookup |> select(lookup_type, lookup_num, description),
by = c("lookup_type", "measurement_value" = "lookup_num")
) |>
left_join(d_occ_lookup, by = c("net_uuid", "species_id", "life_stage")) |>
mutate(
eventID = occ_eventID,
measurementID = as.character(ichthyo_uuid),
measurementType = "abundance by life stage",
measurementTypeID = NA_character_,
meas_val = tally,
measurementValueID = NA_character_,
measurementUnit = "individuals",
measurementMethod = "https://oceanview.pfeg.noaa.gov/CalCOFI/calcofi_info.html",
measurementRemarks = description
) |>
left_join(measurement_unit_vocab, by = "measurementUnit") |>
select(
eventID,
occurrenceID,
measurementID,
measurementType,
measurementTypeID,
measurementValue = meas_val,
measurementValueID,
measurementUnit,
measurementUnitID,
measurementMethod,
measurementRemarks
)
cat(glue("eMoF stage abundance: {nrow(d_emof_stage)}"), "\n")
```
### 10c. Body length measurements
From `ichthyo` rows where `measurement_type = 'size'`:
```{r}
#| label: emof-body-length
d_emof_length <- d_ichthyo |>
filter(measurement_type == "size") |>
left_join(d_occ_lookup, by = c("net_uuid", "species_id", "life_stage")) |>
filter(!is.na(measurement_value)) |>
mutate(
eventID = occ_eventID,
measurementID = as.character(ichthyo_uuid),
measurementType = "body length",
measurementTypeID = NA_character_,
meas_val = measurement_value,
measurementValueID = NA_character_,
measurementUnit = "millimeters",
measurementMethod = "https://oceanview.pfeg.noaa.gov/CalCOFI/calcofi_info.html",
measurementRemarks = paste0("Count: ", tally, "; Total length measurement")
) |>
left_join(measurement_unit_vocab, by = "measurementUnit") |>
select(
eventID,
occurrenceID,
measurementID,
measurementType,
measurementTypeID,
measurementValue = meas_val,
measurementValueID,
measurementUnit,
measurementUnitID,
measurementMethod,
measurementRemarks
)
cat(glue("eMoF body length: {nrow(d_emof_length)}"), "\n")
```
### Combine All eMoF Records
```{r}
#| label: combine-emof
d_emof <- bind_rows(
d_emof_net,
d_emof_stage |> select(-measurementValueID),
d_emof_length |> select(-measurementValueID)
)
cat("eMoF counts by source:\n")
cat(glue(" Net-level: {nrow(d_emof_net)}"), "\n")
cat(glue(" Stage abundance: {nrow(d_emof_stage)}"), "\n")
cat(glue(" Body length: {nrow(d_emof_length)}"), "\n")
cat(glue(" Total: {nrow(d_emof)}"), "\n")
# verify all eMoF records link to a valid eventID or occurrenceID
event_ids <- d_event$eventID
occ_ids <- d_occurrence_out$occurrenceID
emof_with_event <- d_emof |> filter(!is.na(eventID))
emof_with_occ <- d_emof |> filter(!is.na(occurrenceID))
emof_orphan_event <- emof_with_event |> filter(!eventID %in% event_ids)
emof_orphan_occ <- emof_with_occ |> filter(!occurrenceID %in% occ_ids)
if (nrow(emof_orphan_event) > 0) {
warning(glue("{nrow(emof_orphan_event)} eMoF records have invalid eventID"))
}
if (nrow(emof_orphan_occ) > 0) {
warning(glue(
"{nrow(emof_orphan_occ)} eMoF records have invalid occurrenceID"
))
}
```
## Write DarwinCore Archive Files
Export the event core and extension files as CSV:
```{r}
#| label: export-dwc
dir.create(dir_out, recursive = TRUE, showWarnings = FALSE)
write_csv(d_event, file.path(dir_out, "event.csv"), na = "")
write_csv(d_occurrence_out, file.path(dir_out, "occurrence.csv"), na = "")
write_csv(d_emof, file.path(dir_out, "extendedMeasurementOrFact.csv"), na = "")
cat("Wrote CSV files to:", dir_out, "\n")
```
## Create meta.xml
Generate `meta.xml` by mapping CSV column names to DarwinCore term URIs:
```{r}
#| label: create-meta-xml
# DwC term mappings ----
dwc_terms <- list(
event = list(
rowType = "http://rs.tdwg.org/dwc/terms/Event",
idField = "eventID",
terms = c(
eventID = "http://rs.tdwg.org/dwc/terms/eventID",
parentEventID = "http://rs.tdwg.org/dwc/terms/parentEventID",
eventType = "http://rs.tdwg.org/dwc/terms/eventType",
eventDate = "http://rs.tdwg.org/dwc/terms/eventDate",
samplingProtocol = "http://rs.tdwg.org/dwc/terms/samplingProtocol",
sampleSizeValue = "http://rs.tdwg.org/dwc/terms/sampleSizeValue",
sampleSizeUnit = "http://rs.tdwg.org/dwc/terms/sampleSizeUnit",
eventRemarks = "http://rs.tdwg.org/dwc/terms/eventRemarks",
habitat = "http://rs.tdwg.org/dwc/terms/habitat",
footprintWKT = "http://rs.tdwg.org/dwc/terms/footprintWKT",
decimalLatitude = "http://rs.tdwg.org/dwc/terms/decimalLatitude",
decimalLongitude = "http://rs.tdwg.org/dwc/terms/decimalLongitude",
geodeticDatum = "http://rs.tdwg.org/dwc/terms/geodeticDatum",
coordinateUncertaintyInMeters = "http://rs.tdwg.org/dwc/terms/coordinateUncertaintyInMeters",
locationID = "http://rs.tdwg.org/dwc/terms/locationID",
countryCode = "http://rs.tdwg.org/dwc/terms/countryCode",
waterBody = "http://rs.tdwg.org/dwc/terms/waterBody",
datasetID = "http://rs.tdwg.org/dwc/terms/datasetID"
)
),
occurrence = list(
rowType = "http://rs.tdwg.org/dwc/terms/Occurrence",
idField = "occurrenceID",
coreIdField = "eventID",
terms = c(
occurrenceID = "http://rs.tdwg.org/dwc/terms/occurrenceID",
eventID = "http://rs.tdwg.org/dwc/terms/eventID",
scientificName = "http://rs.tdwg.org/dwc/terms/scientificName",
scientificNameID = "http://rs.tdwg.org/dwc/terms/scientificNameID",
kingdom = "http://rs.tdwg.org/dwc/terms/kingdom",
occurrenceStatus = "http://rs.tdwg.org/dwc/terms/occurrenceStatus",
organismQuantity = "http://rs.tdwg.org/dwc/terms/organismQuantity",
organismQuantityType = "http://rs.tdwg.org/dwc/terms/organismQuantityType",
lifeStage = "http://rs.tdwg.org/dwc/terms/lifeStage",
preparations = "http://rs.tdwg.org/dwc/terms/preparations",
basisOfRecord = "http://rs.tdwg.org/dwc/terms/basisOfRecord",
modified = "http://purl.org/dc/terms/modified"
)
),
extendedmeasurementorfact = list(
rowType = "http://rs.iobis.org/obis/terms/ExtendedMeasurementOrFact",
idField = "measurementID",
coreIdField = "eventID",
terms = c(
eventID = "http://rs.tdwg.org/dwc/terms/eventID",
occurrenceID = "http://rs.tdwg.org/dwc/terms/occurrenceID",
measurementID = "http://rs.tdwg.org/dwc/terms/measurementID",
measurementType = "http://rs.tdwg.org/dwc/terms/measurementType",
measurementTypeID = "http://rs.iobis.org/obis/terms/measurementTypeID",
measurementValue = "http://rs.tdwg.org/dwc/terms/measurementValue",
measurementUnit = "http://rs.tdwg.org/dwc/terms/measurementUnit",
measurementUnitID = "http://rs.iobis.org/obis/terms/measurementUnitID",
measurementMethod = "http://rs.tdwg.org/dwc/terms/measurementMethod",
measurementRemarks = "http://rs.tdwg.org/dwc/terms/measurementRemarks"
)
)
)
# helper functions ----
create_field_elements <- function(csv_file, term_map, coreid_field = NULL) {
col_names <- names(read_csv(csv_file, n_max = 0, show_col_types = FALSE))
field_elements <- map(seq_along(col_names), function(i) {
col <- col_names[i]
if (!is.null(coreid_field) && col == coreid_field) {
return(NULL)
}
term <- term_map$terms[[col]]
if (!is.null(term)) {
glue(' <field index="{i-1}" term="{term}"/>')
} else {
message(
"Warning: Column '",
col,
"' in ",
csv_file,
" has no DwC mapping."
)
NULL
}
})
paste(compact(field_elements), collapse = "\n")
}
get_coreid_index <- function(csv_file, coreid_field) {
col_names <- names(read_csv(csv_file, n_max = 0, show_col_types = FALSE))
if (length(coreid_field) > 1) {
coreid_field <- intersect(coreid_field, col_names)[1]
}
which(col_names == coreid_field) - 1
}
# generate meta.xml ----
generate_meta_xml <- function(dir_out) {
event_file <- file.path(dir_out, "event.csv")
event_fields <- create_field_elements(event_file, dwc_terms$event)
event_id_idx <- which(
names(read_csv(event_file, n_max = 0, show_col_types = FALSE)) ==
dwc_terms$event$idField
) -
1
occ_file <- file.path(dir_out, "occurrence.csv")
occ_fields <- create_field_elements(
occ_file,
dwc_terms$occurrence,
coreid_field = dwc_terms$occurrence$coreIdField
)
occ_coreid_idx <- get_coreid_index(occ_file, dwc_terms$occurrence$coreIdField)
emof_file <- file.path(dir_out, "extendedMeasurementOrFact.csv")
emof_fields <- create_field_elements(
emof_file,
dwc_terms$extendedmeasurementorfact,
coreid_field = dwc_terms$extendedmeasurementorfact$coreIdField
)
emof_coreid_idx <- get_coreid_index(
emof_file,
dwc_terms$extendedmeasurementorfact$coreIdField
)
glue(
'<?xml version="1.0" encoding="UTF-8"?>
<archive xmlns="http://rs.tdwg.org/dwc/text/"
xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance"
xsi:schemaLocation="http://rs.tdwg.org/dwc/text/ http://rs.tdwg.org/dwc/text/tdwg_dwc_text.xsd">
<core encoding="UTF-8" fieldsTerminatedBy="," linesTerminatedBy="\\n"
fieldsEnclosedBy=\'"\' ignoreHeaderLines="1" rowType="{dwc_terms$event$rowType}">
<files>
<location>event.csv</location>
</files>
<id index="{event_id_idx}" />
{event_fields}
</core>
<extension encoding="UTF-8" fieldsTerminatedBy="," linesTerminatedBy="\\n"
fieldsEnclosedBy=\'"\' ignoreHeaderLines="1" rowType="{dwc_terms$occurrence$rowType}">
<files>
<location>occurrence.csv</location>
</files>
<coreid index="{occ_coreid_idx}" />
{occ_fields}
</extension>
<extension encoding="UTF-8" fieldsTerminatedBy="," linesTerminatedBy="\\n"
fieldsEnclosedBy=\'"\' ignoreHeaderLines="1" rowType="{dwc_terms$extendedmeasurementorfact$rowType}">
<files>
<location>extendedMeasurementOrFact.csv</location>
</files>
<coreid index="{emof_coreid_idx}" />
{emof_fields}
</extension>
</archive>'
)
}
meta_xml <- generate_meta_xml(dir_out)
writeLines(meta_xml, file.path(dir_out, "meta.xml"))
cat("Generated meta.xml\n")
cat(meta_xml)
```
## Create EML Metadata
Build EML document with geographic, temporal, and taxonomic coverage:
```{r}
#| label: build-eml
# geographic coverage from net sites ----
geo_net <- tbl_site |>
summarise(
lon_min = min(longitude, na.rm = TRUE),
lon_max = max(longitude, na.rm = TRUE),
lat_min = min(latitude, na.rm = TRUE),
lat_max = max(latitude, na.rm = TRUE)
) |>
collect()
geo_coverage <- tibble(
westBoundingCoordinate = geo_net$lon_min,
eastBoundingCoordinate = geo_net$lon_max,
southBoundingCoordinate = geo_net$lat_min,
northBoundingCoordinate = geo_net$lat_max
)
# temporal coverage from tow dates ----
temp_net <- tbl_tow |>
summarise(
tmin = min(time_start, na.rm = TRUE),
tmax = max(time_start, na.rm = TRUE)
) |>
collect()
temp_coverage <- list(
beginDate = as.character(as.Date(temp_net$tmin)),
endDate = as.character(as.Date(temp_net$tmax))
)
# taxonomic coverage ----
d_taxa <- tbl_taxon |> collect()
taxa_coverage <- tbl_species |>
mutate(
worms_id = case_when(
is.na(worms_id) | worms_id == 0 ~ NA_integer_,
TRUE ~ worms_id
)
) |>
collect() |>
filter(!is.na(worms_id)) |>
distinct(species_id, worms_id, common_name) |>
left_join(
d_taxa |> distinct(taxonID, taxonRank, scientificName),
by = join_by(worms_id == taxonID)
) |>
group_by(worms_id, taxonRank, scientificName) |>
summarize(
species_id = first(species_id),
common_name = paste(unique(na.omit(common_name)), collapse = ", "),
.groups = "drop"
) |>
arrange(species_id) |>
filter(!is.na(scientificName)) |>
pmap(function(
species_id,
taxonRank,
scientificName,
common_name,
worms_id,
...
) {
item <- list(
id = glue("calcofi_species_{species_id}"),
taxonRankName = taxonRank,
taxonRankValue = scientificName,
taxonId = list(
provider = "https://www.marinespecies.org",
as.character(worms_id)
)
)
if (!is.na(common_name) && common_name != "") {
item$commonName <- common_name
}
item
})
# build EML document ----
my_eml <- list(
packageId = dataset_id,
system = "calcofi.io",
dataset = list(
shortName = dataset_short_name,
title = dataset_title,
creator = list(
list(
individualName = list(
salutation = "Dr.",
givenName = "Ed",
surName = "Weber"
),
organizationName = "NOAA SWFSC",
electronicMailAddress = "ed.weber@noaa.gov",
userId = list(
directory = "https://orcid.org/",
userId = "0000-0002-0942-434X"
)
),
list(
individualName = list(
salutation = "Phd",
givenName = "Benjamin",
surName = "Best"
),
organizationName = "Ocean Metrics LLC",
positionName = "Marine Data Scientist",
address = list(country = "US"),
phone = "+1(805)705-9770",
electronicMailAddress = "ben@oceanmetrics.io",
userId = list(
directory = "https://www.linkedin.com/profile/view?id=",
userId = "ben-best"
)
)
),
pubDate = format(Sys.Date(), "%Y-%m-%d"),
language = "eng",
abstract = dataset_abstract,
keywordSet = list(
list(
keyword = dataset_keywords,
keywordThesaurus = "GCMD Science Keywords"
),
list(
keyword = "Samplingevent",
keywordThesaurus = "GBIF Dataset Type Vocabulary: http://rs.gbif.org/vocabulary/gbif/dataset_type_2015-07-10.xml"
),
list(
keyword = "Observation",
keywordThesaurus = "GBIF Dataset Subtype Vocabulary: http://rs.gbif.org/vocabulary/gbif/dataset_subtype.xml"
)
),
intellectualRights = intellectual_rights,
licensed = list(
licenseName = "Creative Commons Zero v1.0 Universal",
url = "https://spdx.org/licenses/CC0-1.0.html",
identifier = "CC0-1.0"
),
distribution = list(
scope = "document",
online = list(
url = list(
`function` = "information",
url = dataset_url
)
)
),
coverage = list(
geographicCoverage = list(
geographicDescription = "California Current Large Marine Ecoregion",
boundingCoordinates = geo_coverage
),
temporalCoverage = list(
rangeOfDates = list(
beginDate = list(calendarDate = temp_coverage$beginDate),
endDate = list(calendarDate = temp_coverage$endDate)
)
),
taxonomicCoverage = list(
generalTaxonomicCoverage = "Marine ichthyoplankton (fish eggs and larvae)",
taxonomicClassification = taxa_coverage
)
),
maintenance = list(
description = list(para = ""),
maintenanceUpdateFrequency = "asNeeded"
),
contact = list(
individualName = list(
salutation = "Dr.",
givenName = "Ed",
surName = "Weber"
),
organizationName = "NOAA SWFSC",
positionName = "Research Fish Biologist",
address = list(
deliveryPoint = "8901 La Jolla Shores Dr",
city = "La Jolla",
administrativeArea = "CA",
postalCode = "92037",
country = "US"
),
phone = "(858) 546-5676",
electronicMailAddress = "ed.weber@noaa.gov",
userId = list(
directory = "https://orcid.org/",
userId = "0000-0002-0942-434X"
)
),
methods = list(
methodStep = list(
description = list(para = sampling_methods_description)
),
sampling = list(
studyExtent = list(
description = list(para = study_extent_description)
),
samplingDescription = list(
para = sampling_description
)
)
),
project = list(
title = dataset_title,
personnel = list(
individualName = list(givenName = "Ed", surName = "Weber"),
role = "Data Manager"
),
funding = list(para = funding_information)
)
)
)
eml_xml <- file.path(dir_out, "eml.xml")
write_eml(my_eml, eml_xml)
# replace license placeholder with actual license xml
readLines(eml_xml) |>
str_replace(fixed(names(license_xml)), license_xml) |>
writeLines(eml_xml)
# inject additionalMetadata with GBIF logo
doc <- read_xml(eml_xml)
ns <- xml_ns(doc)
additional_meta <- read_xml(glue(
'
<additionalMetadata>
<metadata>
<gbif>
<dateStamp>{format(Sys.time(), "%Y-%m-%dT%H:%M:%S+00:00")}</dateStamp>
<hierarchyLevel>dataset</hierarchyLevel>
<resourceLogoUrl>{dataset_logo_url}</resourceLogoUrl>
</gbif>
</metadata>
</additionalMetadata>'
))
xml_add_child(doc, additional_meta)
write_xml(doc, eml_xml)
validation_result <- eml_validate(eml_xml)
if (!validation_result) {
cat("EML validation errors:\n")
print(attr(validation_result, "errors"))
stop("EML validation failed")
} else {
cat("EML is valid!\n")
}
```
## Data Quality Checks
Validate event hierarchy, orphan records, and output summary statistics:
```{r}
#| label: check-data-quality
# check for missing WoRMS IDs
d_missing_worms <- tbl_species |>
filter(is.na(worms_id) | worms_id == 0) |>
select(species_id, scientific_name) |>
collect()
if (nrow(d_missing_worms) > 0) {
cat("WARNING:", nrow(d_missing_worms), "species missing WoRMS IDs:\n")
print(d_missing_worms, n = 20)
}
# summary statistics ----
n_cruises <- d_event |> filter(eventType == "cruise") |> nrow()
n_nets <- d_event |> filter(eventType == "net_sample") |> nrow()
n_occs <- nrow(d_occurrence_out)
n_species <- n_distinct(d_occurrence_out$scientificName)
n_emof <- nrow(d_emof)
glue(
"
=== Dataset Summary ===
Total events: {prettyNum(nrow(d_event), big.mark = ',')}
- Cruises: {prettyNum(n_cruises, big.mark = ',')}
- Net samples: {prettyNum(n_nets, big.mark = ',')}
Total occurrences: {prettyNum(n_occs, big.mark = ',')}
Unique species: {prettyNum(n_species, big.mark = ',')}
Total eMoF records: {prettyNum(n_emof, big.mark = ',')}"
)
```
## Package DarwinCore Archive
Create the final DwC-A zip file:
```{r}
#| label: create-dwc-archive
zip_file <- file.path(
dirname(dir_out),
paste0("ichthyo_", Sys.Date(), ".zip")
)
zip(
zip_file,
files = file.path(
dir_out,
c(
"event.csv",
"occurrence.csv",
"extendedMeasurementOrFact.csv",
"meta.xml",
"eml.xml"
)
),
flags = "-j"
)
cat("\nDarwin Core Archive created:", zip_file, "\n")
```
## Sync to Google Cloud Storage
```{r}
#| label: sync-to-gcs
#| eval: false
gcs_bucket <- "calcofi-db"
gcs_publish_prefix <- "publish/ichthyo"
sync_results <- sync_to_gcs(
local_dir = dir_out,
gcs_prefix = gcs_publish_prefix,
bucket = gcs_bucket
)
```
## Validate with obistools
```{r}
#| label: obistools-validate
# read back the CSVs
d_event <- read_csv(file.path(dir_out, "event.csv"))
d_occ <- read_csv(file.path(dir_out, "occurrence.csv"))
d_emof <- read_csv(file.path(dir_out, "extendedMeasurementOrFact.csv"))
# occ.eventID in event.eventID
stopifnot(all(d_occ$eventID %in% d_event$eventID))
# event hierarchy check
cat("=== Event hierarchy check ===\n")
event_check <- check_eventids(d_event)
if (nrow(event_check) == 0) {
cat("PASS: all event parent-child relationships valid\n")
} else {
cat("ISSUES found:\n")
print(event_check)
}
# occurrence → event linkage
cat("\n=== Occurrence-event linkage ===\n")
occ_check <- check_extension_eventids(d_event, d_occ)
if (nrow(occ_check) == 0) {
cat("PASS: all occurrences link to valid events\n")
} else {
cat("ISSUES found:\n")
print(occ_check)
}
# eMoF → event linkage
cat("\n=== eMoF-event linkage ===\n")
emof_event_check <- d_emof |>
filter(!is.na(eventID)) |>
filter(!eventID %in% d_event$eventID)
if (nrow(emof_event_check) == 0) {
cat("PASS: all eMoF eventIDs link to valid events\n")
} else {
cat(nrow(emof_event_check), "eMoF records with invalid eventID\n")
}
# eMoF → occurrence linkage
emof_occ_check <- d_emof |>
filter(!is.na(occurrenceID)) |>
filter(!occurrenceID %in% d_occ$occurrenceID)
if (nrow(emof_occ_check) == 0) {
cat("PASS: all eMoF occurrenceIDs link to valid occurrences\n")
} else {
cat(nrow(emof_occ_check), "eMoF records with invalid occurrenceID\n")
}
# summary by event type
cat("\n=== Events by type ===\n")
d_event |> count(eventType) |> print()
cat("\n=== eMoF by measurementType ===\n")
d_emof |> count(measurementType, sort = TRUE) |> print(n = 50)
```
## Cleanup
```{r}
#| label: cleanup
dbDisconnect(con, shutdown = TRUE)
cat("Database connection closed.\n")
```