---
title: "Ingest CCE-LTER ZooScan PRPOOS"
calcofi:
target_name: ingest_cce_lter_zooscan
workflow_type: ingest
dependency:
- ingest_swfsc_ichthyo
output: data/parquet/cce-lter_zooscan/manifest.json
provider: cce-lter
dataset: zooscan
workflow_url: https://calcofi.io/workflows/ingest_cce-lter_zooscan.html
questions_file: metadata/cce-lter/zooscan/questions.csv
dataset_meta:
dataset_name: ZooScan PRPOOS Zooplankton
description: >
Zooplankton abundance, carbon biomass, and size (Feret diameter, individual
carbon content) from ZooScan optical imaging of CalCOFI / CCE-LTER PRPOOS
net tows on lines 80, 87 and 90, 2005-present, machine-classified into 23
bioclasses (copepod groups, euphausiids, chaetognaths, appendicularians,
salps, doliolids, pteropods, rhizaria and more). One row per
(sample, taxon, measurement_type); per-station tows. Source: SIO Ocean
Informatics ZooScan portal (Ohman Lab; interface by Marina Frants).
citation_main: ""
citation_others: "Plankton sample analysis supported by NSF grants to M.D. Ohman and the CCE-LTER site."
link_calcofi_org: "https://calcofi.org/data/marine-ecosystem-data/zooplankton/"
link_data_source: "https://oceaninformatics.ucsd.edu/zooscandb/"
link_others: []
coverage_temporal: 2005-07 to 2026-04
coverage_spatial: "CalCOFI lines 80, 87, 90 (Southern California)"
license: ""
pi_names: Mark D. Ohman; Marina Frants
tables_owned:
- {table: zooscan_sample}
- {table: zooscan_measurement}
- {table: zooscan_taxon}
- {table: measurement_type, shared: true, note: "shared registry across datasets"}
erd:
color: "#c2e8f0"
editor_options:
chunk_output_type: console
---
## Overview
**Source**: [ZooScan](https://oceaninformatics.ucsd.edu/zooscandb/) — the SIO
Ocean Informatics ZooScan database (Mark Ohman Lab; interface by Marina Frants).
The portal's bulk download is disabled, so `libs/download_zooscan.R` scripts its
public login + PRPOOS plot CGI (`/cgi-bin/tssubplot_new.py`), whose returned
Plotly page embeds the underlying per-station values as a `data:text/csv` URI.
Each bioclass is fetched in both plot modes and consolidated to
`zooscan_prpoos.csv` (see the Acquire step + `by_taxon/_PROVENANCE.md`).
- **Provider**: `cce-lter` (Ohman Lab / CCE-LTER; sibling of `cce-lter_zoodb` and
`cce-lter_euphausiids`).
- **Grain**: one row per **(sample × taxon × measurement_type)**;
`measurement_value = 0` means imaged-but-absent.
- **Samples**: ZooScan-imaged PRPOOS net tows — one per station occupation on
CalCOFI lines 80/87/90, 2005-present.
- **Taxa** (23): ZooScan machine-classified bioclasses (19 WoRMS-resolved; 4
non-taxonomic operational classes — eggs, multiples, nauplii, others).
- **Measurements**: `zooscan_abundance` (No./m²), `zooscan_biomass_carbon`
(mg C/m²), `zooscan_feret_diameter` (mm), `zooscan_carbon_individual` (µg C).
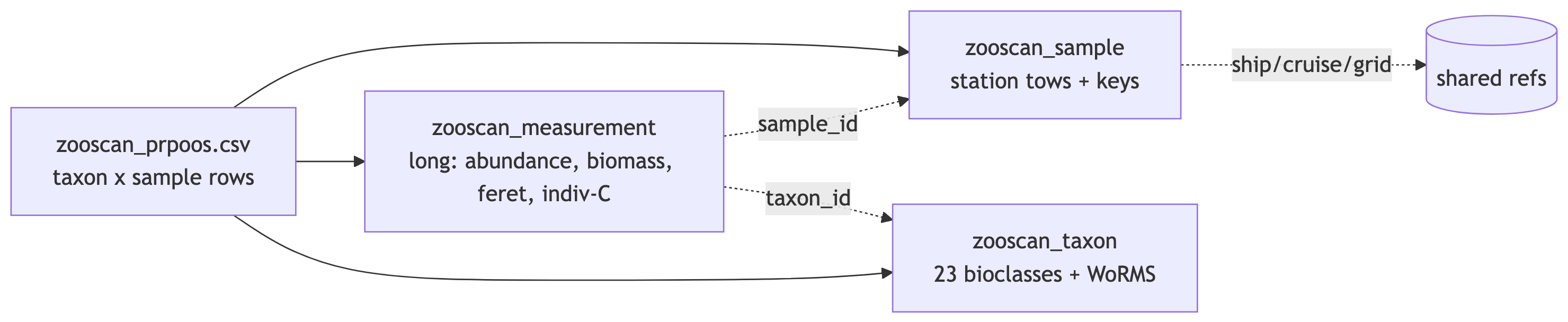
```{mermaid}
graph LR
A[zooscan_prpoos.csv<br/>taxon x sample rows] --> B[zooscan_sample<br/>station tows + keys]
A --> C[zooscan_measurement<br/>long: abundance, biomass, feret, indiv-C]
A --> D[zooscan_taxon<br/>23 bioclasses + WoRMS]
C -.sample_id.-> B
C -.taxon_id.-> D
B -.ship/cruise/grid.-> E[(shared refs)]
```
## Setup
```{r}
#| label: setup
#| message: false
devtools::load_all(here::here("../calcofi4db"))
devtools::load_all(here::here("../calcofi4r"))
librarian::shelf(
CalCOFI/calcofi4db, CalCOFI/calcofi4r,
DBI, dplyr, DT, fs, glue, here, janitor, jsonlite, knitr, lubridate, purrr,
readr, sf, stringr, tibble, tidyr, units, quiet = T)
options(readr.show_col_types = F)
options(DT.options = list(scrollX = TRUE))
source(here("libs/ingest.R")) # overwrite, overwrite_all, dir_data
cc <- read_calcofi_meta(here("ingest_cce-lter_zooscan.qmd"))
provider <- cc$provider
dataset <- cc$dataset
tables_owned <- cc$tables_owned
dir_label <- glue("{provider}_{dataset}")
dir_parquet <- here(glue("data/parquet/{dir_label}"))
db_path <- here(glue("data/wrangling/{dir_label}.duckdb"))
if (overwrite) {
if (file_exists(db_path)) file_delete(db_path)
if (file_exists(paste0(db_path, ".wal"))) file_delete(paste0(db_path, ".wal"))
if (dir_exists(paste0(db_path, ".tmp"))) dir_delete(paste0(db_path, ".tmp"))
}
dir_create(dirname(db_path))
con <- get_duckdb_con(db_path)
load_duckdb_extension(con, "spatial")
meas_type_csv <- here("metadata/measurement_type.csv")
d_meas_type <- read_csv(meas_type_csv)
```
## Acquire Source Data
`libs/download_zooscan.R` scripts the ZooScan portal (public login + PRPOOS plot
CGI) for the 23 bioclasses × 2 plot modes and consolidates the embedded
per-station CSVs into `zooscan_prpoos.csv`. It runs once then caches; set
`overwrite_all = TRUE` in `libs/ingest.R` to re-scrape.
```{r}
#| label: acquire
source(here("libs/download_zooscan.R"))
zs_dir <- path_expand(glue("{dir_data}/cce-lter/ZooScan"))
zs_csv <- download_zooscan(zs_dir, overwrite = overwrite_all)
```
## Read + Clean
The cruise code is `YYYYMM` + a 2-letter ship code (e.g. `200507NH` = Jul 2005,
R/V New Horizon). We parse `year`/`month`/`ship_key` from it, build `site_key`
from line + station, and a `sample_key` per station tow.
```{r}
#| label: clean
zs_dir <- path_expand(glue("{dir_data}/cce-lter/ZooScan"))
# archive source (consolidated + per-taxon extracts) to GCS for provenance
sync_to_gcs(
local_dir = zs_dir,
gcs_prefix = glue("archive/{provider}/{dataset}"),
bucket = "calcofi-files-public",
exclude = c(".DS_Store", "*.tmp", "*.gdoc"))
d_raw <- read_csv(zs_csv)
num <- function(x) suppressWarnings(as.numeric(trimws(as.character(x))))
d <- d_raw |>
transmute(
taxon_slug = taxon,
cruise_orig = trimws(cruise),
line = num(line),
station = num(station),
latitude = num(latitude),
longitude = num(longitude),
max_depth_m = num(max_depth_m),
min_depth_m = num(min_depth_m),
cruise_mid_date = as.Date(cruise_mid_date),
station_date = as.Date(station_date),
local_time_pst = trimws(local_time_pst),
day_night = trimws(day_night),
abundance_per_m2 = num(abundance_per_m2),
biomass_mgC_per_m2 = num(biomass_mgC_per_m2),
feret_diameter_mm = num(feret_diameter_mm),
carbon_content_indiv = num(carbon_content_indiv)) |>
mutate(
# ZooScan reports longitude as positive degrees West — flip to negative (°E)
longitude = -abs(longitude),
year = as.integer(str_sub(cruise_orig, 1, 4)),
month = as.integer(str_sub(cruise_orig, 5, 6)),
ship_key = str_sub(cruise_orig, 7),
site_key = if_else(
is.na(line) | is.na(station), NA_character_,
sprintf("%05.1f %05.1f", line, station)),
datetime_local_pst = suppressWarnings(
ymd_hm(paste(station_date, local_time_pst), quiet = TRUE)),
sample_key = paste(cruise_orig, line, station, station_date, sep = "|"))
cat(glue(
"Read {format(nrow(d), big.mark=',')} (sample x taxon) rows; ",
"{n_distinct(d$sample_key)} samples; {n_distinct(d$taxon_slug)} taxa; ",
"lines {paste(sort(unique(d$line)), collapse='/')}; ",
"{min(d$year)}-{max(d$year)}"), "\n")
```
## Build Taxon Table (WoRMS)
`zooscan_taxon` is the 23 bioclasses with WoRMS AphiaIDs + classification,
resolved offline and cached in `metadata/cce-lter/zooscan/taxon_worms.csv`
(19/23 matched; 4 non-taxonomic operational classes, Q03).
```{r}
#| label: taxon
d_worms <- read_csv(here("metadata/cce-lter/zooscan/taxon_worms.csv"))
zooscan_taxon <- d_worms |>
arrange(taxon_slug) |>
transmute(
taxon_id = row_number(),
taxon_zooscan, taxon_slug,
aphia_id = as.integer(aphia_id),
scientific_name = scientific_name_accepted,
rank, taxon_status = status, taxon_note,
kingdom, phylum, class, order_taxon = order, family)
stopifnot("taxon_slug mismatch source vs WoRMS map" =
all(unique(d$taxon_slug) %in% zooscan_taxon$taxon_slug))
dbWriteTable(con, "zooscan_taxon", zooscan_taxon, overwrite = TRUE)
cat(glue("zooscan_taxon: {nrow(zooscan_taxon)} bioclasses ",
"({sum(!is.na(zooscan_taxon$aphia_id))} WoRMS-matched, ",
"{sum(zooscan_taxon$taxon_status=='non-taxonomic')} non-taxonomic [Q03])"), "\n")
```
## Build Sample Table + Resolve Keys
One row per distinct station tow. `ship_key` (the cruise-code suffix) joins the
shared ship registry for `ship_nodc`/`ship_name`; `cruise_key` is
`YYYY-MM-{ship_nodc}` validated against the cruise registry.
```{r}
#| label: sample-keys
# position/depth are sample properties, but the per-class plot CGI leaves them
# NA on some taxa's rows (e.g. gelatinous classes) — recover the (identical)
# non-NA value per sample; identity columns are constant within sample_key
d_sample0 <- d |>
group_by(sample_key) |>
summarize(
cruise_orig = first(cruise_orig), year = first(year), month = first(month),
ship_key = first(ship_key), line = first(line), station = first(station),
site_key = first(site_key), cruise_mid_date = first(cruise_mid_date),
station_date = first(station_date), local_time_pst = first(local_time_pst),
day_night = first(day_night), datetime_local_pst = first(datetime_local_pst),
latitude = mean(latitude, na.rm = TRUE),
longitude = mean(longitude, na.rm = TRUE),
max_depth_m = mean(max_depth_m, na.rm = TRUE),
min_depth_m = mean(min_depth_m, na.rm = TRUE),
.groups = "drop")
stopifnot("sample_key not unique over sample attributes" =
n_distinct(d_sample0$sample_key) == nrow(d_sample0))
load_prior_tables(
con, parquet_dir = here("data/parquet/swfsc_ichthyo"),
tables = c("ship", "cruise", "grid"), geom_tables = c("grid"), as_view = TRUE)
d_ship <- dbGetQuery(con, "SELECT ship_key, ship_nodc, ship_name FROM ship")
valid_ck <- dbGetQuery(con, "SELECT DISTINCT cruise_key FROM cruise")$cruise_key
d_sample <- d_sample0 |>
left_join(d_ship, by = "ship_key") |>
mutate(
cruise_key = if_else(
is.na(ship_nodc), NA_character_,
sprintf("%04d-%02d-%s", year, month, ship_nodc)),
cruise_key = if_else(cruise_key %in% valid_ck, cruise_key, NA_character_)) |>
arrange(cruise_orig, line, station, station_date) |>
mutate(sample_id = row_number(), .before = 1)
sample_map <- d_sample |> select(sample_id, sample_key)
zooscan_sample <- d_sample |>
select(sample_id, cruise_orig, cruise_key, ship_key, ship_name,
line, station, site_key, latitude, longitude,
max_depth_m, min_depth_m, cruise_mid_date, station_date,
local_time_pst, datetime_local_pst, day_night)
dbWriteTable(con, "zooscan_sample", zooscan_sample, overwrite = TRUE)
n_ship <- sum(!is.na(d_sample$ship_name))
n_cruise <- sum(!is.na(d_sample$cruise_key))
cat(glue(
"zooscan_sample: {nrow(zooscan_sample)} samples; ",
"ship {n_ship}/{nrow(d_sample)} ({round(100*n_ship/nrow(d_sample),1)}%), ",
"cruise_key {n_cruise}/{nrow(d_sample)} ({round(100*n_cruise/nrow(d_sample),1)}%)"), "\n")
```
## Add Spatial
```{r}
#| label: spatial
add_point_geom(con, "zooscan_sample", lon_col = "longitude", lat_col = "latitude")
grid_stats <- assign_grid_key(con, "zooscan_sample")
grid_stats |> datatable(caption = "Grid assignment")
```
## Pivot Measurements to Long Format
The four ZooScan metrics pivot into long form, joined to `sample_id` (via
`sample_key`) and `taxon_id` (via `taxon_slug`). Explicit zeros (imaged-but-
absent) are retained; only NA / non-finite values are dropped.
```{r}
#| label: measurement
meas_recode <- c(
abundance_per_m2 = "zooscan_abundance",
biomass_mgC_per_m2 = "zooscan_biomass_carbon",
feret_diameter_mm = "zooscan_feret_diameter",
carbon_content_indiv = "zooscan_carbon_individual")
zooscan_measurement <- d |>
left_join(sample_map, by = "sample_key") |>
left_join(zooscan_taxon |> select(taxon_id, taxon_slug), by = "taxon_slug") |>
select(sample_id, taxon_id, all_of(names(meas_recode))) |>
pivot_longer(cols = all_of(names(meas_recode)),
names_to = "src_col", values_to = "measurement_value") |>
filter(!is.na(measurement_value), is.finite(measurement_value)) |>
mutate(measurement_type = unname(meas_recode[src_col])) |>
arrange(sample_id, taxon_id, measurement_type) |>
transmute(measurement_id = row_number(),
sample_id, taxon_id, measurement_type, measurement_value)
dbWriteTable(con, "zooscan_measurement", zooscan_measurement, overwrite = TRUE)
cat(glue(
"zooscan_measurement: {format(nrow(zooscan_measurement), big.mark=',')} rows ",
"({format(sum(zooscan_measurement$measurement_value==0), big.mark=',')} explicit zeros)"), "\n")
```
## Add Measurement Types
```{r}
#| label: add-measurement-type
zs_types <- tibble(
measurement_type = c("zooscan_abundance", "zooscan_biomass_carbon",
"zooscan_feret_diameter", "zooscan_carbon_individual"),
description = c(
"Zooplankton areal abundance from ZooScan optical imaging, by image-classified bioclass.",
"Estimated zooplankton carbon biomass from ZooScan optical imaging, by bioclass.",
"Mean Feret diameter (organism size) from ZooScan optical imaging, by bioclass.",
"Mean individual carbon content from ZooScan optical imaging, by bioclass."),
units = c("count/m2", "mgC/m2", "mm", "ugC"),
is_canonical = c(TRUE, TRUE, NA, NA),
`_source_column` = c("abundance_per_m2", "biomass_mgC_per_m2",
"feret_diameter_mm", "carbon_content_indiv"),
`_source_table` = "zooscan_measurement",
`_source_datasets` = "cce-lter_zooscan",
`_qual_column` = NA_character_, `_prec_column` = NA_character_)
new_types <- zs_types |> filter(!measurement_type %in% d_meas_type$measurement_type)
if (nrow(new_types) > 0) {
d_meas_type <- bind_rows(d_meas_type, new_types)
write_csv(d_meas_type, meas_type_csv)
cat(glue("Added measurement type(s): {paste(new_types$measurement_type, collapse=', ')}"), "\n")
} else cat("zooscan measurement types already registered\n")
dbWriteTable(con, "measurement_type", d_meas_type, overwrite = TRUE)
```
## Load Dataset Metadata
```{r}
#| label: load-dataset-metadata
d_dataset <- ingest_yaml_to_dataset_df(read_ingest_yaml(here()))
dbWriteTable(con, "dataset", d_dataset, overwrite = TRUE)
cat(glue("dataset: {nrow(d_dataset)} dataset(s) registered"), "\n")
```
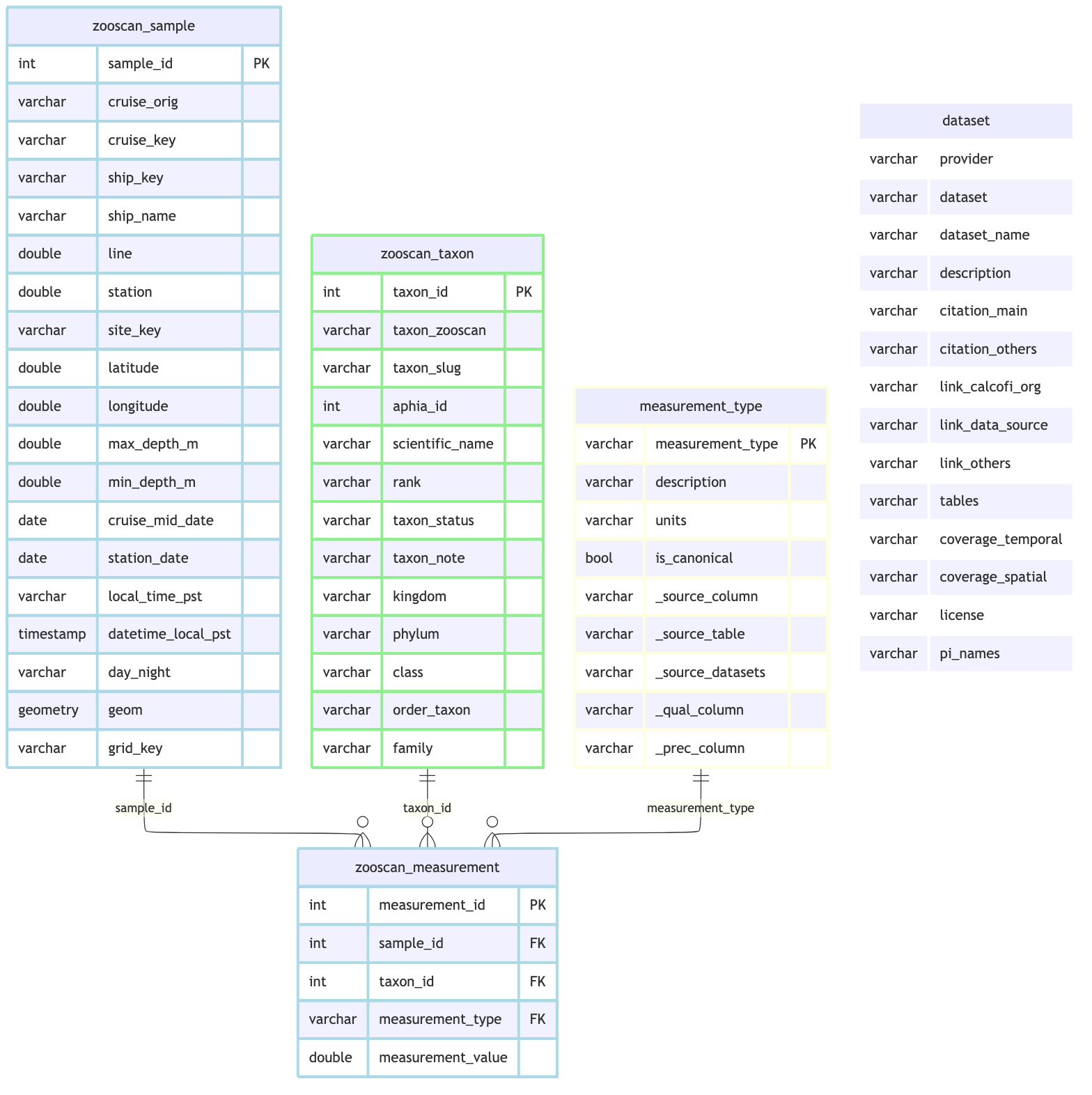
## Schema Documentation
```{r}
#| label: schema
zooscan_rels <- list(
primary_keys = list(
zooscan_sample = "sample_id",
zooscan_measurement = "measurement_id",
zooscan_taxon = "taxon_id",
measurement_type = "measurement_type"),
foreign_keys = list(
list(table = "zooscan_measurement", column = "sample_id",
ref_table = "zooscan_sample", ref_column = "sample_id"),
list(table = "zooscan_measurement", column = "taxon_id",
ref_table = "zooscan_taxon", ref_column = "taxon_id"),
list(table = "zooscan_measurement", column = "measurement_type",
ref_table = "measurement_type", ref_column = "measurement_type")))
cc_erd(
con,
tables = c("zooscan_sample", "zooscan_measurement", "zooscan_taxon",
"measurement_type", "dataset"),
rels = zooscan_rels,
colors = list(
lightblue = c("zooscan_sample", "zooscan_measurement"),
lightgreen = "zooscan_taxon",
lightyellow = "measurement_type",
white = "dataset"))
build_relationships_json(
rels = zooscan_rels, output_dir = dir_parquet,
provider = provider, dataset = dataset)
```
## Validate
```{r}
#| label: validate
results <- validate_for_release(con, checks = "all", strict = FALSE)
cat("Validation:", ifelse(results$passed, "PASSED", "FAILED"), "\n")
if (length(results$errors) > 0)
cat("Errors:\n", paste("-", results$errors, collapse = "\n"), "\n")
# NULLs in cross-dataset keys (cruise_key for cruises absent from the registry)
# are the EXPECTED unmatched remainder (Q02), not a hard failure.
cov <- dbGetQuery(con,
"SELECT AVG(CASE WHEN ship_key IS NOT NULL THEN 1 ELSE 0 END) ship,
AVG(CASE WHEN cruise_key IS NOT NULL THEN 1 ELSE 0 END) cruise,
AVG(CASE WHEN grid_key IS NOT NULL THEN 1 ELSE 0 END) grid
FROM zooscan_sample")
cat(glue("Match coverage: ship_key {round(100*cov$ship,1)}%, ",
"cruise_key {round(100*cov$cruise,1)}%, grid_key {round(100*cov$grid,1)}%"), "\n")
orphan <- dbGetQuery(con,
"SELECT SUM(CASE WHEN s.sample_id IS NULL THEN 1 ELSE 0 END) AS no_sample,
SUM(CASE WHEN t.taxon_id IS NULL THEN 1 ELSE 0 END) AS no_taxon
FROM zooscan_measurement m
LEFT JOIN zooscan_sample s USING (sample_id)
LEFT JOIN zooscan_taxon t USING (taxon_id)")
cat(glue("Orphan measurements: {orphan$no_sample} w/o sample, {orphan$no_taxon} w/o taxon"), "\n")
```
## Data Preview
```{r}
#| label: preview-sample
samp_cols <- dbGetQuery(con,
"SELECT column_name FROM information_schema.columns
WHERE table_name='zooscan_sample' AND data_type NOT LIKE 'GEOMETRY%'")$column_name
dbGetQuery(con, glue("SELECT {paste(samp_cols, collapse=', ')} FROM zooscan_sample LIMIT 100")) |>
datatable(caption = "zooscan_sample — first 100 rows", rownames = FALSE, filter = "top")
```
```{r}
#| label: preview-measurement
dbGetQuery(con,
"SELECT m.measurement_id, m.sample_id, x.taxon_zooscan, m.measurement_type, m.measurement_value
FROM zooscan_measurement m JOIN zooscan_taxon x USING (taxon_id)
ORDER BY m.measurement_id LIMIT 100") |>
datatable(caption = "zooscan_measurement — first 100 rows", rownames = FALSE)
```
```{r}
#| label: preview-taxon
dbGetQuery(con,
"SELECT t.taxon_id, t.taxon_zooscan, t.scientific_name, t.aphia_id, t.rank,
t.taxon_note, COUNT(DISTINCT m.sample_id) AS n_samples
FROM zooscan_taxon t LEFT JOIN zooscan_measurement m USING (taxon_id)
GROUP BY ALL ORDER BY t.taxon_id") |>
datatable(caption = "zooscan_taxon — 23 bioclasses", rownames = FALSE)
```
## Write Parquet Outputs
```{r}
#| label: write-parquet
dir_create(dir_parquet)
mismatches <- list(
measurement_types = collect_measurement_type_mismatches(
con, here("metadata/measurement_type.csv")),
cruise_keys = collect_cruise_key_mismatches(con, "zooscan_sample"))
parquet_stats <- write_parquet_outputs(
con = con,
output_dir = dir_parquet,
tables = c("zooscan_sample", "zooscan_measurement", "zooscan_taxon",
"measurement_type", "dataset"),
sort_by = list(zooscan_measurement = "sample_id"),
strip_provenance = FALSE,
mismatches = mismatches)
parquet_stats |> mutate(file = basename(path)) |> select(-path) |>
datatable(caption = "Parquet export statistics")
```
## Write Metadata
```{r}
#| label: write-metadata
d_tbls_rd <- read_csv(here("metadata/cce-lter/zooscan/tbls_redefine.csv"))
d_flds_rd <- read_csv(here("metadata/cce-lter/zooscan/flds_redefine.csv"))
metadata_path <- build_metadata_json(
con = con,
d_tbls_rd = d_tbls_rd,
d_flds_rd = d_flds_rd,
metadata_derived_csv = here("metadata/cce-lter/zooscan/metadata_derived.csv"),
output_dir = dir_parquet,
tables = c("zooscan_sample", "zooscan_measurement", "zooscan_taxon"),
set_comments = TRUE,
provider = provider,
dataset = dataset,
workflow_url = cc$workflow_url,
tables_owned = tables_owned)
```
## Upload to GCS
```{r}
#| label: upload-gcs
sync_to_gcs(
local_dir = dir_parquet,
gcs_prefix = glue("ingest/{dir_label}"),
bucket = "calcofi-db")
```
## Questions for Data Providers
Follow-up questions for CCE-LTER (Mark Ohman, Marina Frants), tracked in
`metadata/cce-lter/zooscan/questions.csv`.
```{r}
#| label: provider-questions
read_csv(here(glue("metadata/{provider}/{dataset}/questions.csv"))) |>
arrange(factor(priority, c("blocker", "high", "normal", "low")), id) |>
select(priority, question, context, related_field, status) |>
datatable(
caption = "Questions for data providers (ranked by importance)",
options = list(dom = "t", pageLength = 20), rownames = FALSE)
```
## Cleanup
```{r}
#| label: cleanup
close_duckdb(con)
cat(glue("Parquet outputs written to: {dir_parquet}"), "\n")
```
::: {.callout-caution collapse="true"}
## Session Info
```{r session_info}
devtools::session_info()
```
:::